

24-year-old male with neurofibromatosis type 1 who presents with right foot pain and multiple deformities

Presented by H. Thomas Temple, M.D.
July 2021
(Touch to View)

(Touch to View)
View Studies
![]()
View Studies
![]()
(Click to View)
(Click to View)
History
This is a 24-year-old male with neurofibromatosis type 1 who presents with right foot pain and multiple deformities. There are 2 large soft tissue masses, one in the retro Achilles space in the medial ankle and the other on the plantar aspect of the foot. A third smaller and painful mass is located on the dorsolateral aspect of the foot. His chief complaint is pain and difficulty with shoe wear. He has pain in the plantar aspect of his foot in a mass that started out as a small nodule and that has grown substantially over 5 months. He also has pain in the hindfoot due to a large medially-based soft tissue mass as well as a painful nodule over the dorsolateral aspect of the foot. He previously worked as a dishwasher but had to stop working because of pain and the inability to wear a shoe.
The patient is accompanied by his father who also has neurofibromatosis. On detailed questioning, the father states that he is the only person in his family to have neurofibromatosis. He emphatically states that neither his mother nor father nor his 2 sisters had neurofibromatosis. The patient is his only child. The father states that he has had multiple neurofibromas and has an especially large mass over the left nondominant distal arm. The patient also has multiple neurofibromas in addition to those in the right foot and a very small subcutaneous nodule in the right upper chest wall. He also has multiple cafe-au-lait spots and axillary freckling. He has had no other lower extremity deformity such as tibial bowing or a history of tibial pseudarthrosis. He has not had spinal problems and has had no difficulty with visual acuity, frequent headaches, or constitutional symptoms.
On physical examination. There were three distinct masses in the foot, a small mobile mass over the dorsal and lateral aspect of the foot that was tender to palpation and in the subcutaneous tissue. A second mass was in the medial hindfoot; it was deep and non-mobile. The third was on the plantar foot. It was deep, fixed and tender to palpation.
Figure 1a,b
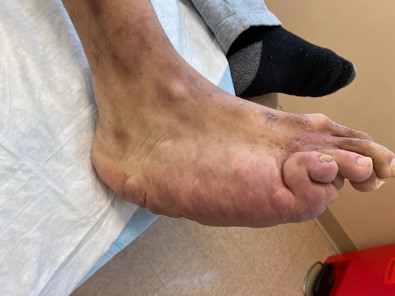
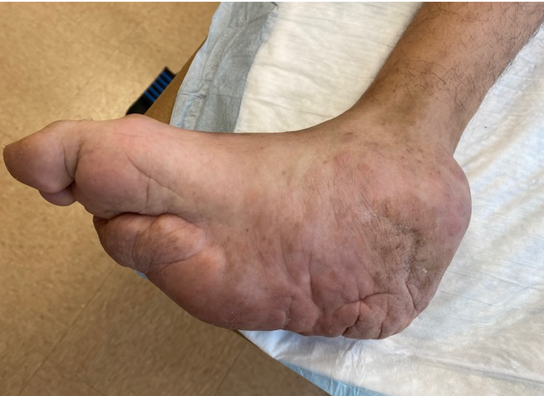
Figure 1a demonstrates the medially based retrocalcaneal mass and the plantar mid foot mass
Figure 1b The small subcutaneous nodule is observed in the dorsal lateral aspect of the foot
Figure 2
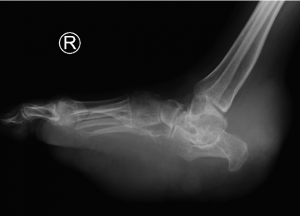
AP, Oblique and Lateral radiographs of the foot show multiple soft tissue densities specifically in the hindfoot (retrocalcaneal region) and the plantar aspect of the foot. In addition, there are erosions in the calcaneus, talus and midfoot.
Figure 3a,b
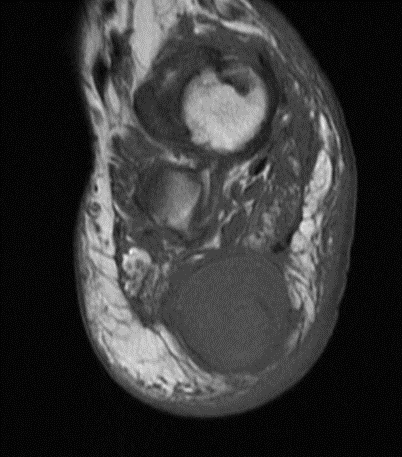
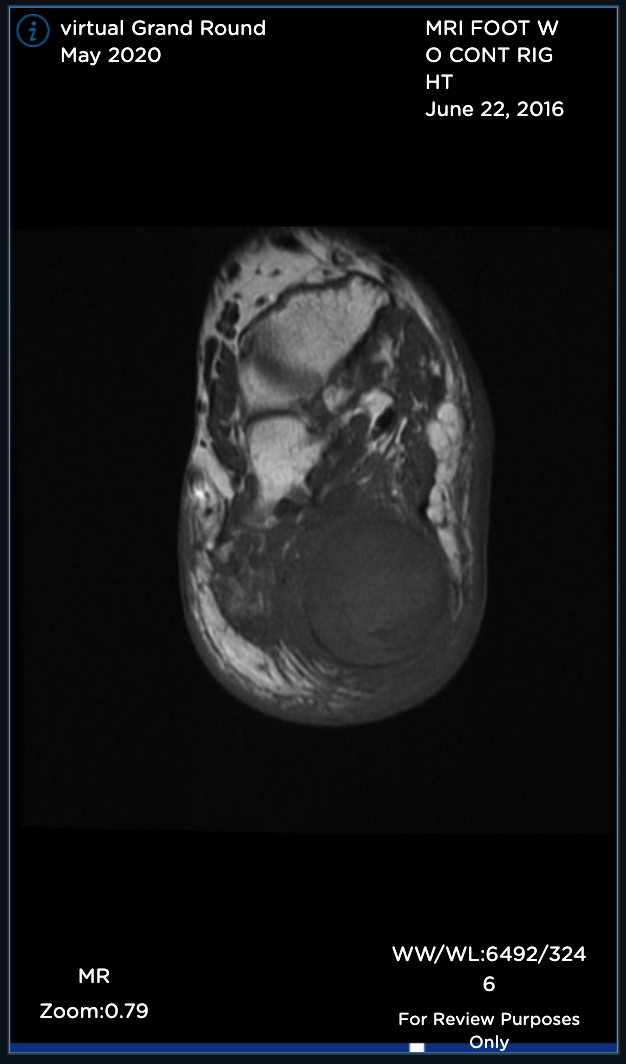
Sagittal MR views both T1 and T2STIR showing both the hindfoot and plantar foot masses. The plantar mass is hyperintense with a nodular area of hypointense signal.
Figure 4a,b
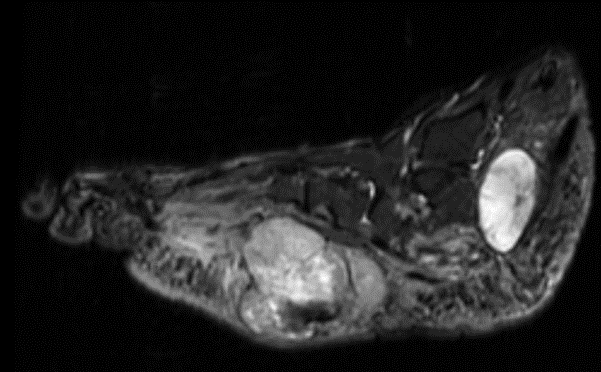
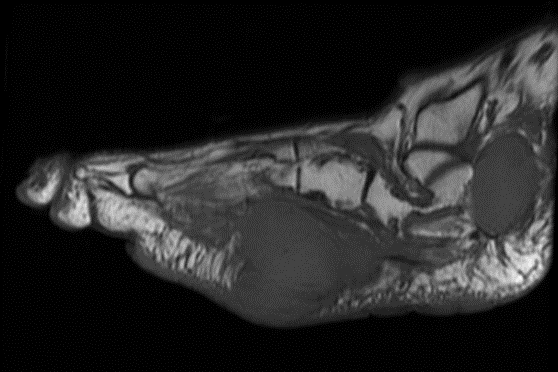
Sagittal MR views both T1 and T2STIR showing both the hindfoot and plantar foot masses. The plantar mass is hyperintense with a nodular area of the hypointense signal.
Click for full MR
Figure 5
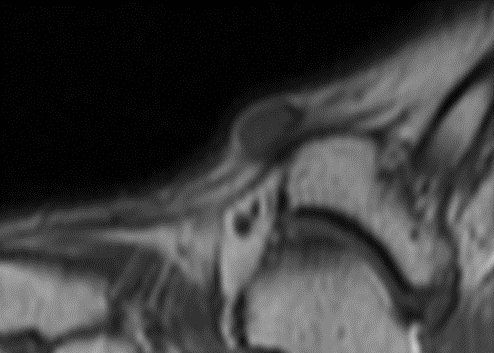
Sagittal T1 MR (TR:647 TE:9.8) demonstrating an ovoid low signal mass with a “string sign” proximally and distally.
Diagnosis
Neurofibromatosis, type I with malignant transformation to of a plexiform neurofibroma.
Operative Technique
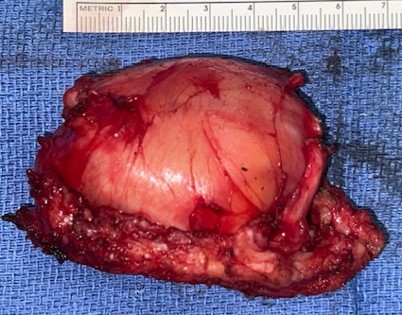
A needle biopsy was performed of the plantar foot mass and multiple cores of tissue were obtained. The histopathologic interpretation was equivocal for malignancy although atypical cells were observed. The patient underwent excision of the hindfoot mass, the small dorsal foot mass and the plantar foot mass. The histopathology of the hindfoot mass was interpreted as a neurofibroma with scattered cells containing atypical nuclei. The tumor was 4.2×2.8×2.6cm and the margins of resection were free with the closest margin being 0.2cm.
Image 1
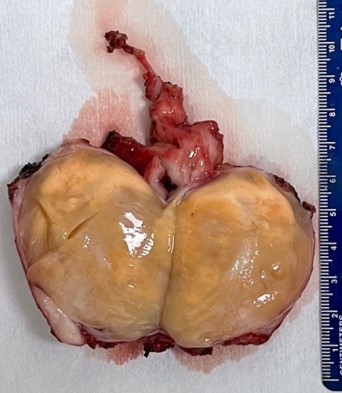
Grossly well-circumscribed tan/whitish mass. The cut surface reveals a relatively homogeneous and was yellow-brown appearing.
Image 2
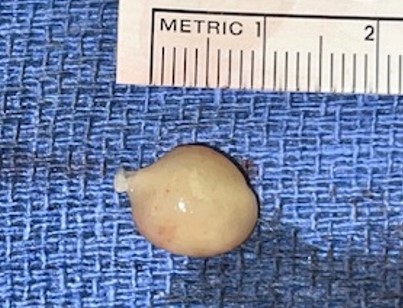
The dorsolateral nodule with a yellow-brown appearance with a well-defined pesudocapsule. This mass was 1.2×1×0.6cm. This lesion had the same histopathologic features as the hindfoot mass.
Image 3

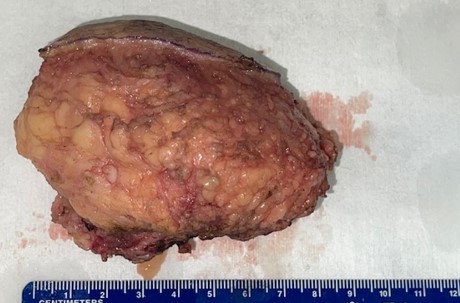
The mass was covered by skin superficially and surrounded by fatty tissue. There was muscle on the deep surface. On cut surface, the mass appeared to be whitish yellow with a firm and rubbery texture.
Image 4a,b
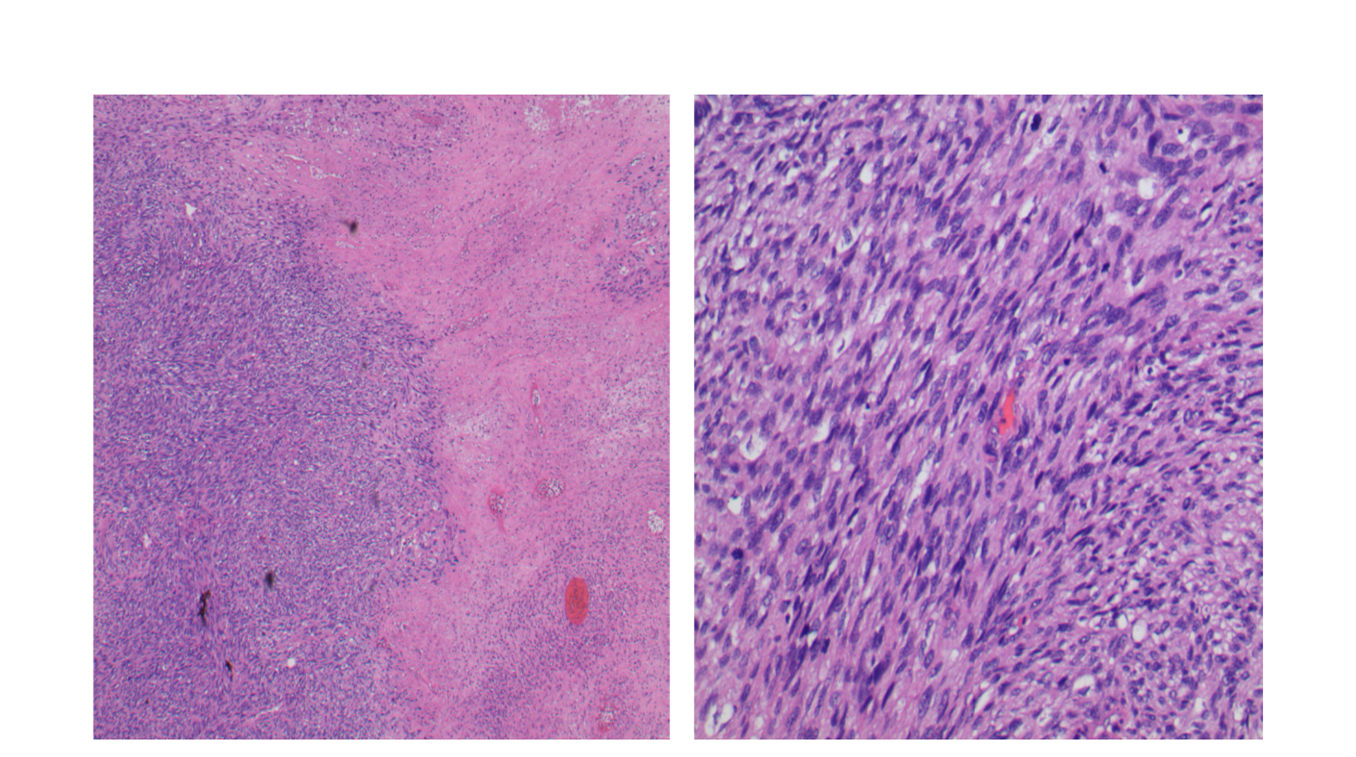
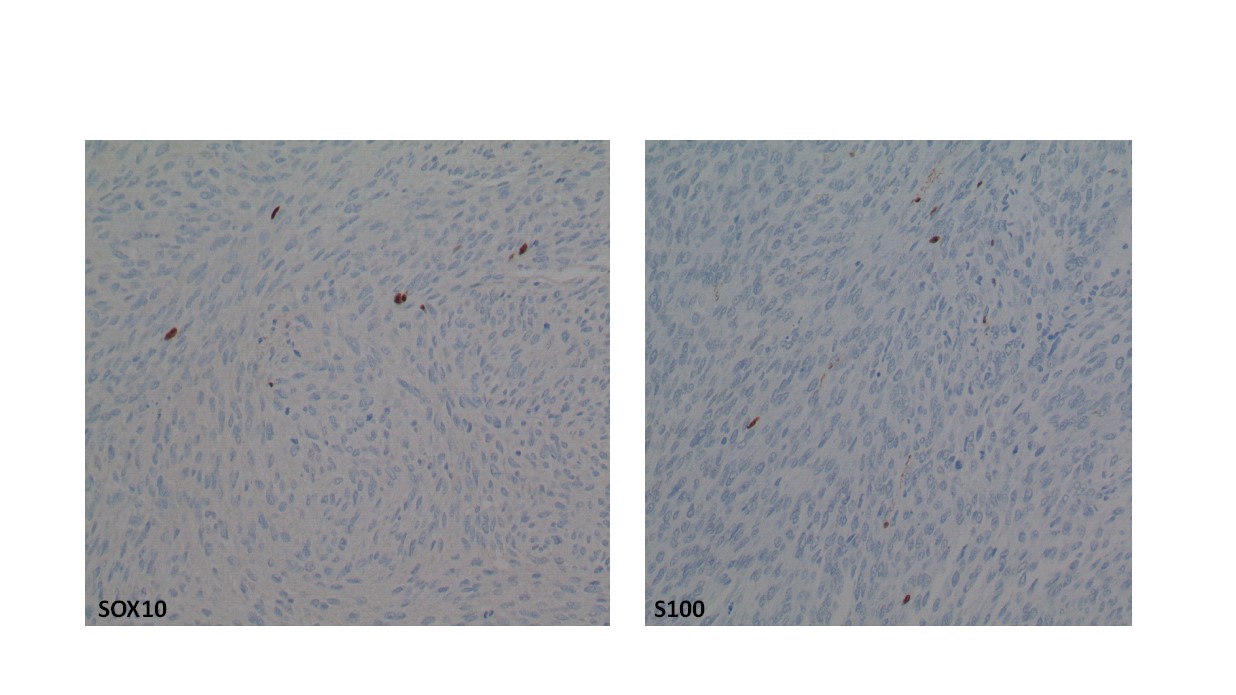
The plantar foot mass revealed a spindle cell sarcoma, grade 3/3. The lesion was cellular (low power, image 4a) with significant nuclear and cellular pleomorphism (high power, image 4b). Mitotic activity was observed. The cells were positive for SOX10 and S-100
image 5a,b
The margins of resection were microscopically positive for tumor (R1). From a functional and oncologic standpoint, the patient opted for a transtibial amputation.
The patient underwent staging to consist of a chest, abdomen and pelvic CT that did not reveal metastatic disease.
Conclusion
This patient with neurofibromatosis, type I had malignant transformation of a large complex neurofibroma on the plantar aspect of his foot.
Peripheral nerves are formed by multiple nerve fascicles separated by “endoneurium”, a fibrous membrane containing fibroblasts and axons protected by Schwann cells. Schwann cells are spindle cells with elongated intertwining cell processes and well-developed basement membranes that provide myelin for the nerve. All components of the nerve sheath have been implicated in the pathogenesis of tumors. The predominant cell type that contributes to each specific tumor is not always obvious and, therefore, the term “peripheral nerve sheath tumor” is used to refer to the entire group.
Neurofibroma and Neurofibromatosis Type 1. Neurofibroma designates a group of benign nerve sheath tumors with a molecular pathogenesis related to loss of function of the NF1 gene. Neurofibromas are most commonly diagnosed in young adults between 20 and 30 years-old with a slight male predominance. Grossly, they are firm, rubbery, gray and glistening. There is a mix of elongated spindle cells and fibroblasts in a background of collagenous fibers with various degrees of cellularity. They can be quite cellular (cellular neurofibroma) and can be confused with dematofibrosarcoma protruberans. The pigmented variant is called a “Bednar tumor”. They are typically S-100 positive with variable CD34 positivity. Neurofibromas can be solitary, diffuse or plexiform. The solitary form occurs in normal individuals whereas the diffuse and plexiform types occur in patients with neurofibromatosis (NF-1), an autosomal dominant condition with near complete penetrance. The NF1 gene is located in s17q11.2. The gene codes for a protein called “neurofibromin” that is believed to inhibit the ras signal transduction pathway. Clinical manifestations of the syndrome include café au lait spots, Lisch nodules, axillary freckling, neurofibromas, tibial pseudarthroses and scoliosis. The café au lait spots have a regular smooth border that is likened to the “Coast of California in contrast to those seen in patients with Albright syndrome where the border is sharp and irregular, resembling the “Coast of Maine”
The treatment of tibial pseudarthroses ranges from a brace or observation to reconstructive surgery or amputation in cases of severe deformity. There are two morphologic patterns of scoliosis: a long C-shaped curve and a short angular curve. The vertebral end-plates are sharp and the neural foramina are widened due to the large dumbbell-shaped neurofibromas arising from the nerve roots The ribs are misshapen like pencils. Most neurofibromas are solitary and sporadic. They affect males and females equally and are usually superficial. They are painless nodules that involve small nerves.
Neurofibromas that are either diffuse or plexiform can be quite disfiguring.
Imaging studies may show heterogeneous uptake with gadolinium administration. Microscopically, the tumor contains spindle cells with wavy elongated nuclei, a myxoid matrix and interspersed “ropey” bundles of collagen fibers. S-100 and neurofilament proteins can be detected by immunohistochemistry. As many as 50% of neurofibrosarcomas (MPNST) arise from neurofibromas in patients with neurofibromatosis, but only 5% of patients with neurofibromatosis develop MPNST. Cellular atypia by itself does not imply malignancy. Pathologists rely more on the presence of high cellularity with increased number of mitoses, and necrosis. Rarely, malignant Triton tumors arise in neurofibromas in patients with neurofibromatosis. This is a tumor with both neurogenic and myogenic (rhabdomyoblastic) differentiation.
Soft tissue sarcomas are a heterogeneous group of malignant tumors that arise from muscles, joints, tendons, nerves and fat. There are two important features that determine how soft tissue sarcomas are treated. These are size and tumor grade. Tumors are sampled and the tissue is prepared onto glass slides so that the pathologist can ascertain whether the tumor has a low risk of spreading to other parts of the body or a high risk. Tumors that are deemed low risk generally are treated with surgery with or without radiation therapy. Tumors that are intermediate or high-grade, depending on their size, greater than or less than 5 cm, are best managed with chemotherapy. The chemotherapeutic agents (Adriamycin and Ifosphamide) for these tumors are very toxic and associated with a number of side effects. The side effects and risks of chemotherapeutic intervention is best discussed by the medical oncologist administering the chemotherapy.
Once the tumor is removed, it is evaluated very carefully by the surgeon and pathologist to ensure that tumor cells do not extend to the resection margin. If tumor does extend to the resection margin and additional tissue can be obtained, the patient undergoes additional surgery. If however tumor extends to margin and just beyond the margin there is a vital structure such as a nerve or artery, this area is generally marked and additional radiotherapy and/or chemotherapy is administered. It should be stated that positive margins do not necessarily result in tumor recurrence and conversely, negative margins do not guarantee tumor free survival.
In our experience, a good response to chemotherapy can improve survival significantly. A bad response however does not portend a poor outcome. With multimodal therapy, local disease control can be achieved in over 90% of patients. Infrequently, negative margins cannot be obtained while attempting to salvage a limb. In these rare circumstances, an amputation may be necessary. In exceptional cases, occasionally a blood vessel and/or nerve can be resected along with the tumor and the artery can be reconstructed and rarely, the nerve can be grafted.
Frequent postoperative surveillance is necessary to ensure that the tumor does not come back at its site of origin or spread to other parts of the body, most notably, the lungs. Patients are surveyed every 3-4 months for the first 2 years then every 6 months up to 5 years then yearly thereafter. We employ ultrasound to assess local disease recurrence. More often than not, the medical oncologist will survey the patient for metastatic disease and this often involves either chest x-rays or more commonly, CT scans of the chest.
For some tumors, it is necessary to assess the lymph nodes as tumor spread can occur in these structures. These tumors include: synovial sarcoma, rhabdomyosarcoma, epithelioid sarcoma and clear cell sarcoma. If preoperative imaging suggests the possibility of lymphatic spread, a sentinel node biopsy may be necessary. This involves sampling of a single lymph node. Lymphatic tumor spread is a negative prognostic factor in overall survival.
Discussion
Want to share your thoughts on this case? Join our discussion on LinkedIn. Click the link below and start commenting on this post!
https://www.linkedin.com/feed/update/urn:li:activity:6786295443493068800
A Note From Dr.Temple
Thank you for taking an interest in my virtual Grand Rounds! My team and I have spent countless hours curating this case, and we couldn’t be more excited to share it with you. mymedicalimages has become a part of my everyday routine, and I encourage you to learn more about this application.
Please be sure to participate in my LinkedIn discussion.
Sincerely,
H. Thomas Temple MD
About mymedicalimages
Hi!
We want to thank you for taking the time to view this month’s virtual Grand Rounds. If this is your first experience with mymedicalimages, we hope it is a pleasant one! If you would like to create a FREE account, just visit mymedicalimages.com or click the menu button or the logo located above and select “Are you a physician?”. Once you sign up, we would like to share with you all the capabilities our application has to offer.
With mymedicalimages, you will never view patient medical images on a CD again! We know the constant struggle of dealing with CDs. Your patients forget them, or they simply don’t work. With the mymedicalimages “Uploader Button” placed onto your physician webpage, or through a safe and secure text or email link, patients can share their images into your account instantly prior to their appointment.
Now that you have easy and secure access to patient images, the possibilities are endless. We go well beyond providing a better way to view, share, and manage medical images, however. With mymedicalimages, you can create a virtual examination room where you can perform telemedicine while viewing DICOM images at your fingertips. Imagine being able to review medical images with patients and telestrate directly on them in order to best communicate findings. With your new account, you have that ability.
Looking to expand your practice? Through our physician profiles, you are able to curate your page allowing yourself to be found quickly and easily for second opinions and consults. Patients and colleagues simply share their images with you.
We know physicians participate in consults daily. However, we also know it isn’t necessarily done the most secure way. With mymedicalimages, you can safely and securely share your images with colleagues from around the world. We break down all barriers by not requiring every party to have a mymedicalimages account, providing collaboration for all. If you find yourself at a conference or want to share images on the fly, you can create a QR code per study. All your colleague has to do is point their camera at it and within seconds they are viewing the same study on their phone.
Now that we have scratched the surface of all your new account has to offer, we hope you take advantage of some, or all, of our features! If you have any questions, our support team is available 24/7 or visit our website at mymedicalimages.com.
If you are interested in being featured in our virtual Grand Rounds, email
vgr@mymedicalimages.com expressing your interest and a team member will get back to you shortly.
All the best,
![]()